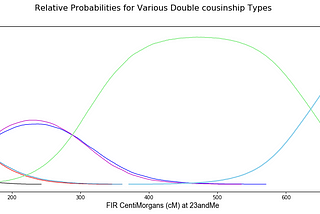
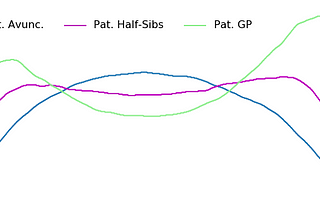
Brit NicholsonDouble Cousin Relationship Predictions with Ped-simDouble cousin predictions are now updated with many more relationship types and a peer-reviewed data sourceMar 9, 2022Mar 9, 2022
Brit NicholsoninAlexandria ScienceA Banner Year for DNA-SCI.comIt will be hard to capture all of the discoveries that have been made and posted at this site in the past twelve months.Oct 19, 2021Oct 19, 2021
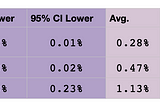
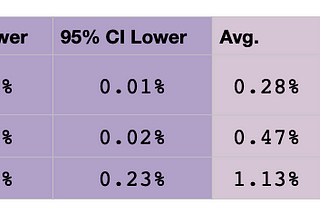
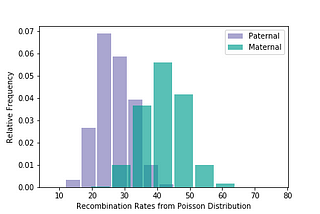
Brit NicholsoninAlexandria ScienceA New Probability Calculator for Genetic GenealogyRelationship predictions are now updated to include differences in maternal and paternal recombination rates as well as validation of…Apr 6, 2021Apr 6, 2021
Brit NicholsonDNA Coverage with Multiple KitsHow much of an ancestor’s or relative’s DNA can you reproduce when you combine kits of multiple testers?Mar 4, 2021Mar 4, 2021
Brit NicholsonWhy does 23andMe show that I share an unusually high amount of DNA (50%) with my full-sibling?Alternate title/misleading answer: 23andMe counts FIR twiceMar 3, 2021Mar 3, 2021
Brit NicholsonHow far back is the ancestor who gave me a particular ethnicity?People often ask a question similar to this one: If my ethnicity report is showing 5% Iberian, how far back should I expect to find an…Mar 3, 2021Mar 3, 2021
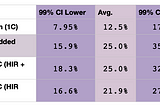
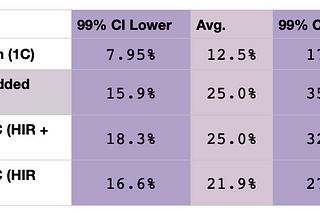
Brit NicholsonCan You Just Add the Averages and Ranges for Double Relationships?People often ask how their DNA match with a cousin will be inflated if they have a double relationship with that cousin. Some helpful…Jan 6, 2021Jan 6, 2021
Brit NicholsonYour Siblings Have DNA Kits, too? Use Them to Their Full Potential.Using very simple math the get the most out of multiple kits.Dec 1, 2020Dec 1, 2020
Brit NicholsoninAlexandria ScienceHow Much of an Ancestor’s or Relative’s DNA Do You Have?After two years of genetic modeling and simulation, I believe I’ve finally arrived at just about the best model I can makeMay 18, 2020May 18, 2020
Brit NicholsoninAlexandria ScienceNew and Improved Autosomal Genetic ModelA comparison of three autosomal models, including results of the new model that predicts the amount of shared DNA between various…May 18, 20201May 18, 20201